



A HybISS-based cell type map reveals specific cellular neighborhoods
We collected tissue samples from six donors targeting five discrete anatomical regions, congruent to the previously described locations of cells in scRNA-seq datasets1,19, and grouped them into three major anatomical regions: trachea (ventral side of the airway with surrounding mesenchyme), proximal lung (generation 2-3 intralobar bronchus with surrounding mesenchyme and occasionally alveoli) and distal lung (distal/terminal bronchioli and alveolar tissue close to the edge of the lobes). After histology-based assessment, two out of six donors were excluded due to multiple signs of pathology, including fibrosis or large immune infiltrations. Samples from the remaining four donors, among which two were smokers, one ex-smoker and one non-smoker (Supplementary Data 1), were subjected to mRNA quality controls to reject the samples with low or diffuse RNA signal (Methods, Supplementary Fig. 1A). High-quality samples from different locations were processed by three different complementary SRT technologies (Fig. 1A, Supplementary Table 1). In order to detect all cell types simultaneously on each tissue section, we first classified cells based on previously published scRNA-seq data (Supplementary Fig. 1B)1,3 and generated a probe panel for HybISS using Spapros, as previously described20. We applied this panel on two selected representative samples using HybISS15. The obtained dataset was used to detect most cell types and states in the trachea and lung. In order to detect particular cell states, we used SCRINSHOT, a more sensitive SRT method, with an additional panel of major cell type markers and genes showing intra-cluster gene expression variation16,20. Finally, a section from each anatomic location was analyzed with an untargeted SRT method Visium in order to validate the probe panels. Visual cross-validation sections of the three methods on serial tissue sections demonstrated consistent marker gene expression patterns, and the performance of targeted methods (Supplementary Fig. 1C).
First, we identified cell types by profiling sections from three anatomic locations of four donors by highly multiplex method HybISS using a gene panel consisting of 162 genes (Supplementary Data 2). After decoding, 157 genes were annotated. Cells were segmented based on DAPI-stained nuclei, and gene transcript signals were assigned to the nearest nuclei21. We excluded cells with low transcript counts and finally processed a total of 260,398 cells for further analysis and clustering based on their expression profiles. This separated the cells into six major classes, assigned according to marker gene expression: airway epithelial, immune, alveolar epithelial, endothelial, stromal, and submucosal gland (SMG) (Supplementary Data 2, 3). The cells in these classes mapped to their expected histological locations (Supplementary Fig. 2A). By further subclustering of each class, we revealed and annotated 28 cell types (Fig. 1B, Supplementary Data 3), corresponding to the majority of the adult lung cell types, described in previous scRNA-seq studies2,4. Based on positivity for corresponding cell type marker genes in the RNA-seq atlases1,2,3, we manually annotated seven additional cell types that could not be assigned by the unsupervised sub-clustering of the HybISS data either due to their sparsely detected gene expression, such as in T lymphocytes, NK cells, a group of T and/or NK cells (here labeled T/NK), and aerocytes, or due to their low abundance, such as ionocytes, tuft cells, rare tuft-like cells, and squamous-like cells (Supplementary Fig. 2B, Supplementary Table 2). The expected marker genes were expressed in the corresponding cell types, except for 11 genes, which were detected in very few cells of the annotated clusters (Supplementary Fig. 2C-D, in brackets). The overall performance of the HybISS marker gene probe panel and cell type annotations were tested using integration mapping22 with the corresponding scRNA-seq dataset1. Cell types detected in HybISS demonstrated high prediction score corresponding to the expected scRNA-seq cell type annotations (Supplementary Fig. 3A). Ten cell types demonstrated prediction scores to more than one annotation, due to their mixed origin and marker co-expression with related types, or due to lack of cell type-specific markers. These cell types were annotated based on their location and morphology on histological images (Supplementary Fig. 3B). Their annotation and location was confirmed in complementary SCRINSHOT- and Visium-processed serial sections, as described below. In total, our analysis resulted in identification of a total of 35 cell types among 221,130 cells that were mapped onto the tissue topography in situ (Fig. 1B-D). The data were deposited in an open access searchable browser that visualizes cell type annotation, gene expression levels and histological stainings (see Data Availability in viewers for HybISS Atlas).
Complementing the HybISS datasets, we profiled sequential sections by SCRINSHOT16, employing a pre-validated gene panel of 64 marker genes20. This panel overlapped with HybISS by 43 genes. The gene counts per cell were obtained as for HybISS, and after clustering major cell types were assigned according to marker gene positivity (Supplementary Data 4). We annotated 24 cell types by clustering and also defined an additional cell state (AT0) based on previous publications reporting the co-expression of airway and alveolar epithelial markers9 (Supplementary Fig. 4). Two of the cell clusters (immune nan and epithelial nan) could not be further subdivided due to the detection of only general and non-discriminative markers. Seven cell types contained multiple clusters or subclusters that were annotated as subtypes or states (Supplementary Fig. 5). We inspected and confirmed the signals and cell types on the tissue to ensure their expected location. We correlated spatial distribution of cell types in HybISS and SCRINSHOT by plotting them on serial sections and comparing their proportions in the same tissue compartments of serial sections (Supplementary Fig. 6). In addition, we analyzed sequential sections of two tissue blocks using the Visium protocol, followed by the Stereoscope method23 to deconvolve the cell type composition of each spot using the finest annotation from scRNA-seq dataset from Madissoon et al1 as a reference. Visual assessment confirmed that the location of most assigned cell types within the tissue was consistent between all three methods (Supplementary Fig. 7, A-B). Overall cell type proportion distributions across manually defined tissue compartments on serial sections correlated significantly between the three SRT methods (Supplementary Fig. 7C), confirming the specificity of each of the technologies. The combinatorial approach with three methodologies hand-in-hand overcomes limitations of individual spatial mapping protocols, such as cellular resolution or variable detection sensitivity.
Specific cell types and states in distinct tissue compartments
In order to characterize cell type distribution, we defined cell type compositions across tissue locations by calculating the relative frequency of each cell type within each profiled region (Fig. 1C). We treated distal regions as a single location due to their similarity in cellular composition. Several cell types exhibited a regional preference, for example AT1 and AT2 epithelial cells were mostly present in distal lung, whereas B plasma and SMG cells mainly occupied tracheal regions, as expected (Fig. 1C, D). To further dissect the relative spatial distributions of cell types and describe consistent cellular colocalizations across the mature lung, we performed neighborhood proximity enrichment analysis of all datasets. This revealed multiple cellular colocalizations, including most cell types, except tuft and lymphatic endothelial cells. The colocalizations were combined into larger neighborhoods, which were characterized by defined histological features (Fig. 1D, E). In addition to the SMG and airway epithelial neighborhoods, we also revealed a group of cell types in proximity to AT1, AT2 and aerocytes, which included stromal, endothelial, monocytes and NK cells (Fig. 1E). This neighborhood was therefore labeled alveolar parenchyma. A distinct neighborhood composed of adventitial fibroblasts, venous and immune cells, was revealed in both peri-bronchial and peri-SMG locations (Fig. 1D, E). In summary, our combined data provide an overview of 35 cell types and their occurrences in the topographic map of healthy adult lung, defining distinct cellular neighborhoods based on cell type proximity in the entire tissue.
To complement predicted neighborhoods, we addressed common classification of tissue compartments using histologic landmarks and cellular morphology, similarly to a recently published approach24. We related cellular morphologies in hematoxylin-eosin (H&E) staining with cell-type annotations and gene expression on the same section. We defined peri-epithelial, such as (i) the SMG, (ii) airways, and (iii) alveolar compartment, and non-epithelial, such as (iv) veins, (v) arteries, and (vi) cartilage (Supplementary Fig. 8A-C, Fig. 2A, Methods). These histological subdivisions were largely in agreement with the predicted neighborhoods (Figs. 1E, 2A–C) and covered most of the tissue area (Supplementary Fig. 8A-C). We mapped the 35 HybISS-based cell types and SCRINSHOT-based subtypes in relation to histologically defined tissue compartments, assessing compartment-specific gene expression in cell types.

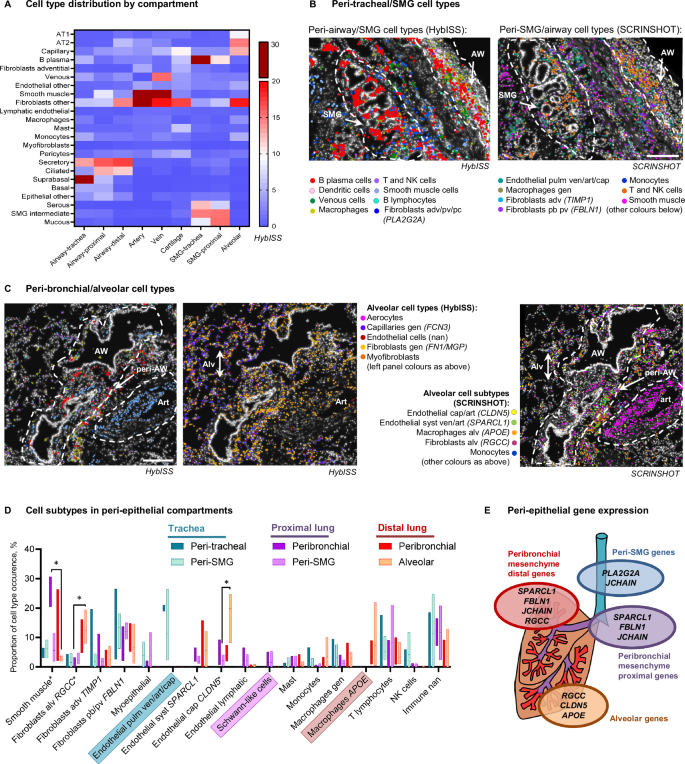
A Heatmap of average (mean) cell type proportions (%) in manually annotated histological compartments using HybISS cell type maps. Proportion calculations include non-annotated cells, not presented in the heatmap. Cell type/subtype maps on top of nuclei (DAPI, white) of HybISS (left) and SCRINSHOT (right) datasets of donor 1 trachea (B) and distal lung (C), respresentative images from four biological replicates. Dotted outlines: peribronchial and airway (AW), submucosal gland (SMG), alveolar (Alv) and arterial (Art) compartments. Cell type abbreviations: pb peribronchial, adv adventitial, alv alveolar, pv perivascular, cap capillary, gen general, art arterial, ven venous, nan not annotated. Scale bar 200 µm. D Graph of mean cell type/subtype proportions in each of the peri-epithelial compartments. Floating bars indicate Min/Max values as bounds of box, with a line on mean proportion per compartment, and indicated in percent (%), n = 3 in tracheal regions, n = 4 in proximal and distal lung regions. Values were compared in lung regions using Friedman two-sided test followed by Dunn’s multiple comparisons test. Significant differences (P < 0.05) are indicated by asterisk (*). Adjusted P values are as follows: P = 0.0370 for fibroblasts (RGCC) between alveolar parenchyma and proximal bronchi; P = 0.0370 for smooth muscle cells between alveolar parenchyma and proximal bronchi; P = 0.0370 for endothelial (CLDN5) cells between alveolar parenchyma and proximal SMG. The cell subtypes that dominated in one anatomic location are highlighted with coloured boxes. E Schematic summary of the gene expression in the parenchyma of each compartment. Source data are provided as a Source Data file.
First, we focused on the submucosal gland structure, which includes a duct protruding from the airway lumen branching into the tubules and acini composed of mucous and serous cells25. SMG mucous and serous cells expressing their corresponding markers (Supplementary Data 3 and 5, Supplementary Fig. 2D, Supplementary Fig. 4G) were either intermingled with each other or were found in continuous patches of either mucous or serous cells. In the ducts, we detected cells expressing airway secretory cell markers (LCN2, ALDH1A3, SCGB3A1) together with serous or rarely mucous markers (Supplementary Fig. 4G) and therefore called these cells SMG intermediate. These cells corresponded to SMG duct cells from scRNA-seq in SCRINSHOT dataset (Supplementary Fig. 4G-H). In contrast, SCGB3A2 was expressed in a subpopulation of serous cells, usually located in small tubules and not in the duct (Supplementary Fig. 5C, Supplementary Fig. 6C)10,26. The previously reported description of an SMG immune niche1, as well as our neighborhood analysis (Fig. 1E) suggests specific cell type enrichment around the gland. The peri-SMG non-epithelial compartment in the trachea and proximal lung was presented by B-plasma cells and fibroblasts (Fig. 2A, B), confirmed in Visium dataset, with JCHAIN (marker of B plasma cells), as well as PLA2G2A (adventitial fibroblast) expression (Supplementary Fig. 9). Cell subtype analysis in the SCRINSHOT dataset indicated the presence of fibroblasts expressing FBLN1, myoepithelial cells, T, NK and other immune cells, which could not be more precisely annotated (nan) (Fig. 2B and D, Supplementary Fig. 6D). Interestingly, endothelial cells around the tracheal SMG predominantly expressed all endothelial markers (SPARCL1, IGFBP7 and CLDN5), potentially corresponding to pulmonary vasculature (venous/arterial/capillary), whereas in the lobes we found either SPARCL1, or IGFPB7 or CLDN5 positive cells potentially corresponding to systemic arterial/venous, lymphatic or capillary cells, respectively (Fig. 2B–D, Supplementary Fig. 5B-D, Supplementary Fig. 10). In conclusion, we identified unexpected heterogeneity of the epithelial cell populations in the SMG, and specific B plasma and adventitial fibroblast cells within its compartment.
As expected from the neighborhood analysis, the peri-airway compartment contained very similar cell type combinations as the peri-SMG one (Fig. 2B–D). However, the peri-bronchial compartments varied in different anatomic locations. For example, we only found histologic ganglia structures with VIM/CD9 double-positive cells (annotated Schwann-like cells according to their morphology) in proximal lung (Fig. 2D, Data viewer for SCRINSHOT Atlas). Additionally, smooth muscle cells were most abundant in proximal bronchi, whereas the distinct populations of endothelial cells expressing either SPARCL1 (which could indicate venous, aerocyte or arterial cells) or CLDN5 (capillary or arterial) were only found in lobular bronchi. In distal lung, APOE expressing macrophages also appeared in peribronchial region (Fig. 2C, D). These data suggest high heterogeneity of gene expression along the proximo-distal axis of the airways.
The alveolar parenchyma was defined by the presence of AT1 and AT2 epithelial cells (Fig. 2A, Supplementary Fig. 8C) and was dominated by capillaries (including aerocytes), and general (non-adventitial) fibroblasts (Fig. 2A, C). In comparison to other compartments, the alveolar parenchyma had the highest proportion of CLDN5 positive endothelial cells, most likely corresponding to alveolar capillaries, and APOE (alveolar) macrophages (Fig. 2C–D)1,2. Fibroblasts positive for RGCC (alveolar fibroblasts) were dominating in the distal lung (Fig. 2D). Fibroblasts in the distal lung also expressed higher FN1 and RGCC, and lower PLA2G2A and C3 levels, compared to the fibroblasts in the other regions (Supplementary Fig. 8D). These gene expression patterns suggest the existence of multiple fibroblast subtypes located in different peri-epithelial tissue compartments. Statistical analysis indicated that some cell types demonstrate significant variability between compartments. However, pairwise comparison test within each anatomical location did not give any significant results, when all cell types were compared simultaneously. We therefore can conclude that the majority of subtype gene expression is driven by anatomical location, rather than by the compartment within each location.
Among non-epithelial compartments, large vessels and cartilage contained endothelial cells expressing most endothelial markers, and FBLN1 positive fibroblasts. The peri-arterial compartment was distinguished by the high proportion of smooth muscle cells (Supplementary Fig. 8E). The peri-venous compartment contained small proportions of all mesenchymal cell types. Chondrocytes were detected only in the Visium dataset (Supplementary Fig. 7A). The peri-chondrial regions were composed of FBLN1 and PLA2G2A positive fibroblasts, and occasionally capillaries, pericytes, and mast cells (Fig. 2A, Supplementary Fig. 8E).
In order to define the differences between SPARCL1 and CLDN5 expression in the vascular system, we first estimated the general distribution of endothelial subtypes in SCRINSHOT and HybISS and defined that most of these cells are located in small vessels or capillaries and are of mixed origin (Supplementary Fig. 10A). Immunofluorescent staining confirmed that the vessels around the airways were surrounded by a thin layer of aSMA, indicating that they belong to venule and/or arteriole category, whereas CLDN5-positive vessels located in the alveolar region lacked aSMA staining, and belong to capillary network (Supplementary Fig. 10B). Therefore we concluded that the airways are surrounded by a network of small venules or arterioles, which are positive for SPARCL1 and low-positive for CLDN5. The cells expressing high levels of CLDN5 are most likely general (non-aerocyte) alveolar capillary cell population.
Location-specific distributions of cell types and cell states with distinct gene expression patterns in different compartments define cell type niches and inform on potential cell-to-cell signaling domains. Our data reveal an enrichment of APOE macrophages and endothelial cells highly expressing CLDN5 in alveoli. Peri-bronchial and peri-SMG regions on the other hand, were composed of FBLN1 fibroblasts, and JCHAIN plasma cells, with PLA2G2A fibroblasts enriched around the gland and cartilage (Fig. 2, Supplementary Fig. 8E), which were also confirmed by our Visium dataset (Supplementary Fig. 9). The definition of regional gene expression variation in non-epithelial cell types, such as fibroblasts, immune and endothelial cells in the healthy lung provides a basis for the precise comparison of the same regions in the diseased states. This may distinguish the regional gene expression variations from the disease-associated ones.
Multiple cell states with distinct topologies in the airway epithelium
We next focused on the topography of cell diversity in the airway epithelium. Among bronchial epithelial cells, a total of 11 cell types were identified, including basal, suprabasal, ciliated, deuterosomal, and neuroendocrine cells, as well as manually assigned ionocytes, tuft (brush), rare tuft-like and squamous-like cells (Fig. 1B, Supplementary Fig. 2B, Supplementary Table 2, Supplementary Data 3). The remaining bronchial epithelial cells were split into two groups. First, secretory cells expressing the AGR2, SFTPB, BPIFB1, MUC5AC markers and comprising 33% of total bronchial epithelial cells and second a smaller group comprising 12% of total bronchial epithelial cells, which were positive for the general epithelial marker gene SLPI (Supplementary Data 3). These cells were found spread along the airways, but also occasionally in SMGs and alveoli. However, they were negative for the characteristic epithelial cell type markers, such as mucins or secretoglobins and were designated not annotated ‘nan’ cells (Fig. 3A, Supplementary Data 3). They could represent less differentiated epithelial cells or unknown cell states.

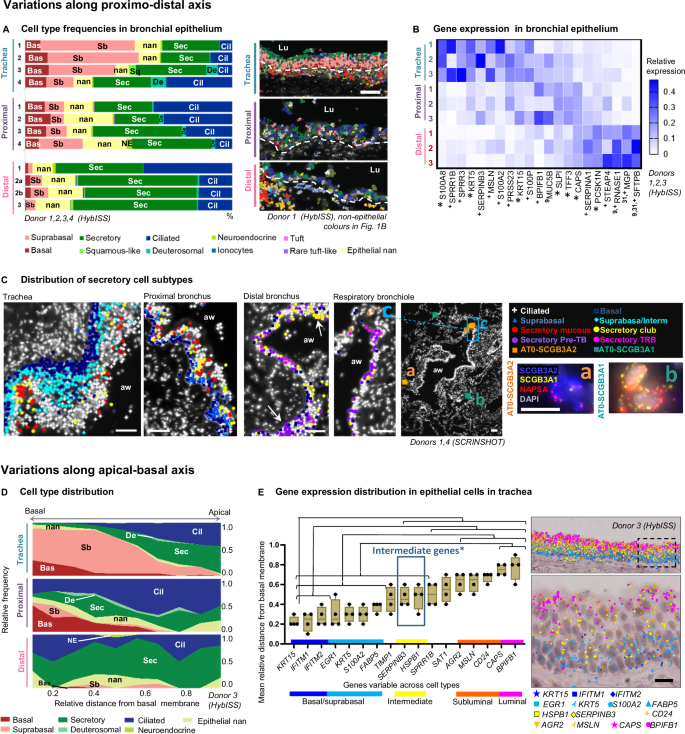
A Cell type frequencies in the airway epithelium according to HybISS. Left: stacked bar plot representing relative frequencies of the airway epithelial cell types across regions from different donors, using samples with representative numbers of airway cells. Right: cell type maps in the indicated regions from donor 1, respresentative images from four biological replicates. Spots represent detected transcripts, colored according to the corresponding cell type of the cell they were assigned to. Colors as in Fig. 1B. Nuclei: gray. Lu lumen, dashed line: approximate location of basal membrane. Scale bar: 50 µm. B Heatmap of the relative mean gene expression in airway epithelium of variable epithelial markers across regions (colors) in three analysed donors (numbers). The expression is normalized by gene, dividing by sum of values in each row. Superscript numbers: references of previous studies, reporting variable expression of the corresponding marker along the proximal-distal axis. Asterisk*: statistically significant expression differences of the corresponding marker between regions (P < 0.05, repeated measures ANOVA with Geisser-Greenhouse correction, followed by Tukey’s multiple comparisons test, all 161 detected genes tested). Plus + : having highest mean change. C Maps of epithelial cell types detected by SCRINSHOT in the indicated regions of the airways, respresentative images from four biological replicates. Arrows: cell clusters. Inserts in respiratory bronchiole map: (a-b) SCRINSHOT images of representative AT0 cells with either SCGB3A2 (a, orange squares) or SCGB3A1 (b, jade squares) dominating expression. (c) Zoomed area of respiratory bronchiole. Nuclei: gray. Scale bar in maps: 50 µm. Scale bar in SCRINSHOT images 10 µm. aw airway. D Area plots representing the relative apical-basal cell type distribution across regions according to HybISS (data from one representative donor 3). X axis: relative distance of cells from the basal membrane. Y axis: relative frequency of cell types. E Mean gene expression of significantly layer-variable markers along the apical-basal axis of the tracheal epithelium (n = 4). Individual data points represent biological replicates. Box bounds indicate standard deviation, line indicates mean, and whiskers indicate minimum and maximum values. Significant differences (P < 0.05) between gene dot distances are calculated using nested one-way ANOVA followed by Tukey’s multiple comparisons test, and indicated with lines and brackets. Exact P values are indicated in Source Data. Blue box* indicates the genes that are located differently from both basal and luminal values. To the right, image of HybISS-detected transcripts in tracheal epithelium, where each transcript is shown as a characteristic coloured shape. The image is representative of four biological replicates. Squared area in the image is shown magnified below. Scale bar 20 µm. Source data are provided as a Source Data file.
To investigate cell type composition diversity along the airway proximal-distal axis, we further characterized the composition of the airway epithelium in tracheal, proximal and distal airway sections from individual donor samples by HybISS. In the trachea, the epithelium was dominated by suprabasal cells, whereas in the intralobar airways the epithelium was mainly composed of secretory and ciliated cells (Fig. 3A). Gene expression comparison across the regions confirmed this distribution, with basal and suprabasal (KRT5, KRT15, S100A2), and squamous (SPRR3/1B) genes expressed predominantly in the trachea. In addition, this analysis revealed further variability across the regions, including, for example, mesothelin (MSLN) expression in trachea, trefoil factor 3 (TFF3) and SLPI in the proximal lung, and surfactant protein B genes (SFTPB) in the distal lung (Fig. 3B). These variable genes could mainly be attributed to distinct secretory cell populations or regional variations in the secretory cell transcriptomes. Statistical analysis of all four donors confirmed a significant dominance of AGR2-positive populations in the trachea compared to other regions, and the higher abundance of SFTPB-positive populations in distal lung, compared to the trachea (Supplementary Fig. 11A). This analysis identifies consistent differences in epithelial composition between three anatomical regions along the proximo-distal axis of the airway tree. To further define the location of the major secretory cell subtypes, we quantified gene expression by SCRINSHOT, targeting characteristic cell type markers for goblet, club and terminal bronchiole epithelial cells (pre-TB or TASC or RAS)10,13,27 in three different locations (Supplementary Data 4, 5). We found club cells in all three anatomical regions but localized goblet cells dominating in trachea and proximal lung and pre-TB cells only in distal lung10,28 (Supplementary Fig. 11B). Similar to HybISS dataset, a group of epithelial cells could not be annotated by characteristic markers and was assigned epithelial nan (Supplementary Fig. 11B-C, Supplementary Data 5). Interestingly, subpopulations of the club cells co-expressed low levels of mucins. Moreover, in contrast to pre-TB cells, the club cells expressed genes encoding antimicrobial proteins (such as LTF, LCN2, and CYP2F1, Supplementary Fig. 5C), suggesting a specialized role in epithelial immunity. Both distal club and pre-TB cells were located in small clusters along distal bronchi and respiratory bronchioles (Fig. 3C). We also detected terminal respiratory bronchiolar (TRB) secretory and alveolar type 0 (AT0) cells in peri-bronchial and alveolar regions respectively (Fig. 3C). AT0 cells were defined by co-expression of the alveolar type II cell marker NAPSA, and low but evident levels of either SCGB3A29, or SCGB3A1 or LCN2, suggesting additional heterogeneity in this cell type (Supplementary Fig. 5C, Fig. 3C). Overall our spatial analysis of cell type locations and distributions confirms the previously published proximo-distal variation in gene expression, and reveals that club cells are present in all anatomical regions at similar proportions, whereas other secretory cell populations are location-specific.
The analysis so far revealed large gene expression heterogeneity in the distal airway epithelium and a dominant abundance of suprabasal epithelial cells in the thicker tracheal epithelium (Fig. 3A–C). To investigate the suprabasal cell type further and define its topological relationships within the pseudostratified tracheal epithelium, we investigated gene expression in relation to the distance from the basal membrane to the lumen. In the HybISS dataset, basal and suprabasal cells were enriched close to the epithelial basement membrane, according to their nuclei locations. In contrast, secretory and ciliated cells were enriched in more apical positions, ciliated cells being closest to the airway lumen (Fig. 3D, Supplementary Fig. 11D). Quantification of the mRNA signals along the distance from the basal membrane to the lumen defined basally-enriched (KRT15, IFITM1 and IFITM2), and apically-enriched mRNAs (BPIFB1, CAPS), as well as an intermediately located gene expression program (SERPINB3 and HSPB1)29 (Fig. 3E). In addition, S100A8 and SPRR1B, SPRR3 were variably expressed in different donors in scattered distances from the basal membrane (Supplementary Fig. 11E-F), which could be related to epithelial condition30. SCRINSHOT analysis of consecutive tracheal sections targeting S100A9 and KRT13 detected their expression in the intermediate and apical layers, and SERPINB3 in the intermediate layer (Supplementary Fig. 11G). Only a subset of S100A9 cells expressed KRT13 (Supplementary Fig. 11G). This supports that the KRT13 positive cells might correspond to hillock cells, a distinct tracheal cell state, in line with recent observations31. Our spatial analysis reveals multiple cell states located in layers of the pseudostratified tracheal epithelium. Overall, there is a strong correspondence to three layers along apical-basal axis of the airway epithelium, with characteristic HSPB1 and SERPINB3 expression in the intermediate layer, distinct from previously characterized hillock and squamous cell states3,7,31, which in this study appear donor- and layer-variable (Supplementary Fig. 11H).
Rare cell type mapping reveals region-specific neuroendocrine cells
The variability of gene expression patterns in the apical epithelial cells along the proximo-distal axis could partly be explained by differential exposure to external factors. Certain environmental factors are sensed by specialized rare cells located in the airways. Ionocytes and tuft cells have been identified in the nasal epithelium and distal airways, whereas neuroendocrine cells were predominantly located in trachea and intermediate airways3. Our HybISS-based analysis allowed mapping most of these cell types. Yet, the expression levels of their markers were low, making it difficult to extract safe conclusions regarding their differential distribution. To explore the potential variation and location of rare cells, we developed a marker panel targeting pulmonary ionocytes, tuft-like cells and neuroendocrine (NE) cells, based on the previously integrated human lung cell atlas from scRNA-seq2, as well as specific airway epithelial cell types3 and embryonic single-cell atlas studies32,33. Since neuroendocrine cells of adult lung are diverse, a precise selection of markers was performed to uncover potential heterogeneity in neuroendocrine cell phenotypes, targeting the four most abundant adult NE genes, as well as two genes marking a NE population discovered predominantly in the developing embryonic lung8,32. We used these markers in SCRINSHOT and located rare epithelial cell types manually by positivity for the expected markers. We assessed gene expression in 180 rare epithelial cells from four donors, and clustered these cells, identifying at least four groups of neuroendocrine cells: (1) NE-GRP, expressing GRP and low levels of ASCL1, (2) NE-ASCL1 expressing ASCL1 and low levels of GRP, (3) NE-GHRL positive for GHRL and CFC1, and (4) NE-PCSK1N expressing variable levels of PCSK1N, GRP and ASCL1 (Fig. 4A, B, Supplementary Fig. 12A-B). All NE groups sparsely expressed variable levels of CHGB and were represented in each donor. GHRL and GRP differential location was confirmed by immunofluorescence (Fig. 4C). ASCL3 expression defined ionocytes, and cells expressing variable levels of POU2F3, RGS13 and CRYM were annotated as tuft cells, the latter including a rare tuft-like cell population expressing previously published markers NREP and HES63 (Fig. 4A, B, Supplementary Fig. 12A-B). We visually analyzed samples from three regions of four donors and selected samples with large parts of the airway (covering a continuous airway length of at least 2 mm per section) for further quantification. In order to create a uniform regional annotation of their positions disregarding the variable epithelial thickness, we quantified rare cell types per length of basal membrane from the selected samples from at least two donors per region (Supplementary Fig. 12C) and then assessed in proportion to the remaining airway epithelial cells (Fig. 4D). Ionocytes were observed in all anatomical locations, preferentially in trachea and proximal bronchi (Fig. 4A, D). Tuft and rare tuft-like cells were mostly located in proximal bronchi, but were observed in other locations along the airway, occasionally in close proximity to other rare cells, but also solitary (Fig. 4A, D). The three neuroendocrine cell identities were observed across locations, but interestingly, GHRL-positive NE cells only appeared in distal bronchioles of three donors and were not observed in trachea or proximal lung (Fig. 4A, D, Supplementary Fig. 12D). GHRL-positive NE cells have previously been detected in embryonic and pediatric datasets and these cells were hypothesized to gradually disappear in adulthood33,34. Our results indicate that targeted spatially-resolved methods allow the detection of low abundant or very rare cell populations with high efficiency, enabling the evaluation of the roles of these cells in lung function.

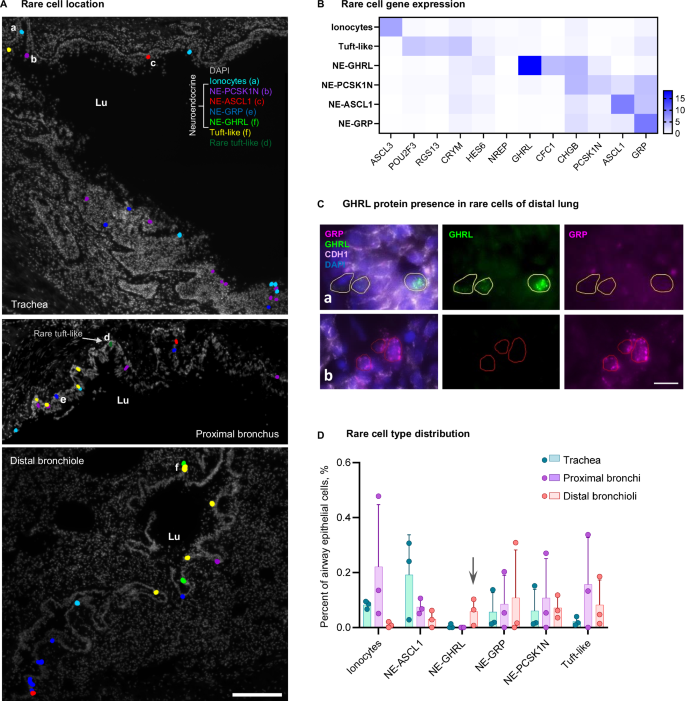
A Maps of rare cell types detected with SCRINSHOT from three anatomical regions (donor 4), respresentative images from three biological replicates. Scale bar 200 µm. Lu lumen. Raw SCRINSHOT signal images of cells labelled with letters (a-f) are shown in Supplementary Fig. 12B. B Heatmap of gene expression demonstrating unique and overlapping marker genes within the detected rare cell types from four donors. At least two out of four donors demonstrated each cell type in each anatomical region, except NE-GHRL population. Number of cells quantified (180): ionocytes—41, tuft-like:—27 (including tuft—24 and rare tuft-like—3), NE-GHRL—9, NE-PCSK1N—32, NE-ASCL1—40, NE-GRP—31. C Immunofluorescent staining for GHRL (green) and GRP (magenta) of two subtypes of neuroendocrine cells, as well as epithelial membrane marker CDH1 (white) on top of nuclei (DAPI, blue), distal lung of donor 4, which had largest number of neuroendocrine cells from three stained samples. Areas (a) and (b) are crops from a larger image in Supplementary Fig 12D. Scale bar: 10 µm. D Bar plot of the number of the detected cells per total airway epithelial cell number (shown in %) from four donors, error bars: standard deviation. Individual data points represent biological replicates (only donors with airway larger than 2 mm of basal membrane length). Arrow: NE-GHRLpos cells appearing only in distal lung. Source data are provided as a Source Data file.
Spatial analysis of early-stage COPD samples demonstrates AT0 cell alterations and new cellular neighborhoods
We further explored the utility of our topographic atlas as a reference to detect deviations in cellular proportions, gene expression and neighborhoods in diseased lung tissue. We focused on chronic obstructive pulmonary disease (COPD), a common lung disease affecting airways and alveoli, using samples from 3 patients with COPD GOLD stage II obtained from the most distal lung locations (corresponding to region 3c in Fig. 1A). These samples were derived from the tumor-free regions from lung cancer surgeries. Two healthy atlas samples together with one histologically normal tumor-free lung sample of a chronic lung disease (CLD)-free cancer patient were processed side-by-side for comparison. Samples contained variable airway sizes (large, medium, small bronchioles and respiratory bronchioles). We applied a modified SCRINSHOT panel to test the expression of the 41 most selective genes in order to define major cell types (Supplementary Fig. 13A).
We annotated 84,631 high-quality cells and defined 20 major cell types according to their markers, regardless of their surroundings (Supplementary Fig. 13B). The major cell classes were equivalently represented in all analyzed samples (Supplementary Fig. 13C), however the proportion of AT1 cells was decreased and the proportion of T lymphocytes increased in COPD samples (Fig. 5A). Previous extensive scRNA-seq studies of COPD samples reported a shift in the expression of epithelial secretory cell gene programs, where proximal airway gene expression levels gradually increased in distal epithelial cells of COPD airways35 leading to a decrease in the proportion of pre-TB (TASC) secretory cells10. Recent publications reported an increase in bronchial secretory cell type marker expression in the AT2 cells from COPD patients27, and an increase in AT0 cell state9. We therefore subclustered both bronchial secretory and AT2 cells and defined a population of cells co-expressing AT2 (NAPSA, SFTPC) and airway (SCGB3A1, SCGB3A2, LTF, LCN2) markers, which we annotated as AT0 cells (Supplementary Fig. 13B, D). We found that the proportion of these AT0 cells was significantly increased in all COPD samples (Fig. 5A, B). Histological compartment analysis indicated that these AT0 cells were mostly in the alveolar regions in proximity to the large and small airways, and in respiratory bronchioles, but the increase primarily affected alveolar regions (Supplementary Fig. 14A-E). This in situ increase in AT0 state is in line with the scRNA-seq analysis arguing for a general upregulation of the proximal secretory cell type program in epithelial cells, not only in the airways, but also in the alveoli27,35.

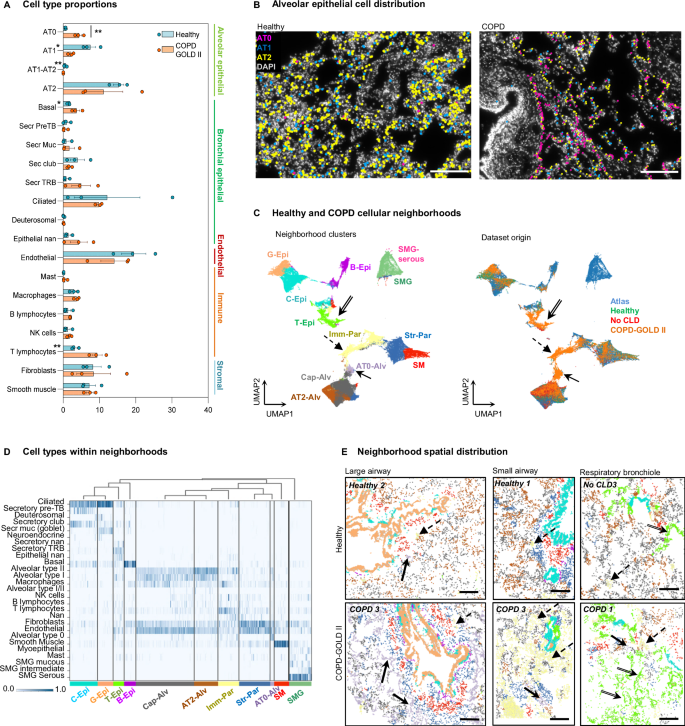
A Box plot of the cell type numbers in healthy and COPD (mean ± standard error, with individual values). Significant differences are highlighted with asterisk (**, adjusted P = 0.0087), according to unpaired parametric two-sided multiple t-test (20) of logit-transformed data with Holm-Sidak correction. Individual data points demonstrate samples from different donors and patients, n = 3. The direct cell type comparison without the correction also revealed significant differences between healthy and COPD samples in the following cell types: AT0 (P = 0.0004), AT1 (P = 0.0127), AT1-AT2 (P = 0.0090), basal cells (P = 0.0295), T lymphocytes (P = 0.0088), labeled by asterisks at the cell type name. B Representative (from three biological replicates) maps of alveolar epithelial cell types detected in healthy and COPD samples, nuclei: gray. Scale bar 200 µm. C UMAP plots of analyzed cells labeled according to their cellular neighborhoods (left) and their corresponding condition (right). Neighborhood annotations: Cap-Alv capillary-enriched alveoli, AT2-Alv AT2-enriched alveoli, AT0-Alv AT0-enriched alveoli, C-Epi club-enriched epithelium, G-Epi goblet-enriched epithelium, T-Epi secretory TRB-enriched epithelium, B-Epi basal cell-enriched epithelium, Imm-Par immune cell-enriched parenchyma, Str-Par stromal cell-enriched parenchyma, SM smooth muscle, SMG submucosal gland, SMG-serous serous cell-enriched submucosal gland, TRB terminal respiratory bronchiole. Arrows indicate the clusters that are predominantly composed of COPD-derived cells. D Heatmap exploring the cell type composition of each neighborhood, with cells (vertical lines) grouped by their assigned neighborhood cluster (x-axis) represented in Fig. 5C (left). Bar color represents the ratio of neighborhood enrichment with each cell type for each cell. E Maps of cell type neighborhoods in healthy and COPD samples in alveolar region with large and small airway and respiratory bronchioles. Color code as in C (left). Scale bar 200 µm. Simple arrows: AT0-enriched alveolar neighborhood, dashed arrows – immune-enriched parenchyma, double-line arrows – secretory TRB-enriched epithelium. Source data are provided as a Source Data file.
We extended our analysis to find potential COPD-specific cellular niches. First, we compared healthy and diseased peri-bronchial and alveolar non-epithelial cells and found the increase in T lymphocytes in both COPD compartments, and NK cells in peri-bronchial, with a decrease in CLDN5 endothelial and RGCC fibroblasts in alveolar compartments (Supplementary Fig. 15A). Following this, we performed neighborhood analysis (Methods)36, and clustered the COPD-cellular neighborhoods together with the ones from the healthy atlas, which contained the coordinates of 218,496 cells, 164,719 of which were grouped into 30 annotated cell types, from three anatomic locations (3-4 donor samples per location). The integrated data separated into twelve neighborhood clusters with two of them corresponding to the SMG, and ten of them matching the distal lung regions (Fig. 5C). Three of these neighborhoods were composed predominantly of cells deriving from COPD samples of all three patients (Supplementary Fig. 15B, Fig. 5C, arrows, Supplementary Fig. 15C, arrows). The first COPD-cell neighborhood (termed T-Epi) located in terminal bronchioles and alveoli, contained cells from all disease-samples expressing TRB and AT0 markers, an unannotated secretory epithelial cell type, AT1 cells and endothelial cells (Fig. 5D). This neighborhood was particularly increased in proportion in one of the patients (Supplementary Fig. 15B, COPD II-1). The second COPD-specific cellular neighborhood (AT0-Alv) was composed of cells expressing AT0, AT1 and AT2 cell markers, fibroblasts and endothelial cells (Fig. 5D). This neighborhood mainly contained cells from another COPD patient (Supplementary Fig. 15B, COPD II-3). Finally, the third COPD-cell neighborhood (Imm-Par) was composed of T lymphocytes and other immune cells, fibroblasts, endothelial and AT2 epithelial cells, and was consistently increased in all three patients analyzed. (Fig. 5D, E, Supplementary Fig. 15B-D). This is in accordance with the known inflammatory nature of the COPD. Moreover, we identified two neighborhoods (AT2-Alv) and (Cap-Alv), composed of alveolar epithelial cells, endothelial cells, macrophages and fibroblasts, which were decreased in all three COPD patients compared to the healthy lung tissue samples (Fig. 5C-E, Supplementary Fig. 15B-D). This is consistent with the onset of alveolar simplification, which is an important component of the COPD pathology. This spatial analysis based on the topography of the healthy lung describes deviations in the cellular locations and neighborhood composition in the diseased lungs. We detected a reduction in the alveolar epithelial neighborhoods (AT2-Alv and Cap-Alv). Instead, AT0 cells in COPD patients were increased and contributed to different COPD-specific neighborhoods, AT0-Alv, T-Epi and Imm-Par (Fig. 5D, E). The neighborhood analysis reveals a consistent shift in the balance of the distal airway and alveolar cell phenotypes. We conclude that the usage of the spatially resolved healthy lung cell atlas as a reference aids the detection of cellular composition and cellular environments of tissue samples derived from diseased lungs.
link